
Todos os cálculos do grupo GFQSI são feitos com o pacote Quantum Espresso que implementa a Teoria do Funcional da Densidade (DFT – Density Functional Theory) em condições de contorno periódica por uma base de onda planas.
-
Cálculos de energia eletrônica e otimização de geometria
Através do método auto-consistente, é possível obter a densidade e a energia eletrônica total de um determinado sistema. Essa densidade poderá ser usada mais tarde para cálculos de pós-processamento e a energia eletrônica também poderá ser usada para cálculos termodinâmicos de transições de fase, adsorções, desidratações e para cálculos de energia de formação. Otimizações de geometria também se mostram bastante necessárias para estudos estruturais e são obtidas pela minimização do gradiente de energia pelo método BFGS (Broyden-Fletcher-Goldfarb-Shanno).
-
Cálculos de fônons em Γ
A determinação dos fônons são sempre importantes para a verificação do mínimo obtido pela otimização de geometria e também para o estudo termodinâmico de diversos processos. Os cálculos de fônons são baseados na aproximação do oscilador harmônico e obtidos pela Teoria da Perturbação do Funcional da Densidade (DFPT – Density-Functional Perturbation Theory). As estimativas das cargas efetivas também nos permite avaliar qual modo é ativo no infravermelho e qual é a sua intensidade de absorção.
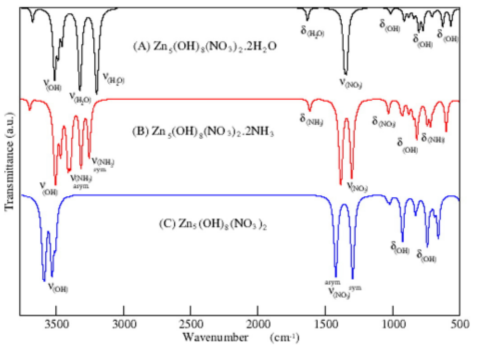
-
Cálculos de Pós-processamento
Como já mencionado acima, a obtenção da densidade nos permite obter diversas informações interessantes como basicidade/acidez e informações sobre as interações existentes em um determinado composto. Os cálculos de pós-processamento mais utilizados no grupo são:
1 – Densidade de Estados (DOS – Density of States) e pDOS (projected Density of States)
Os cálculos de DOS e pDOS nos permite verificar a presença de sítios básicos e ácidos em um cristal ou até mesmo em uma superfície e, até mesmo, comparar cada sítio através de suas projeções.
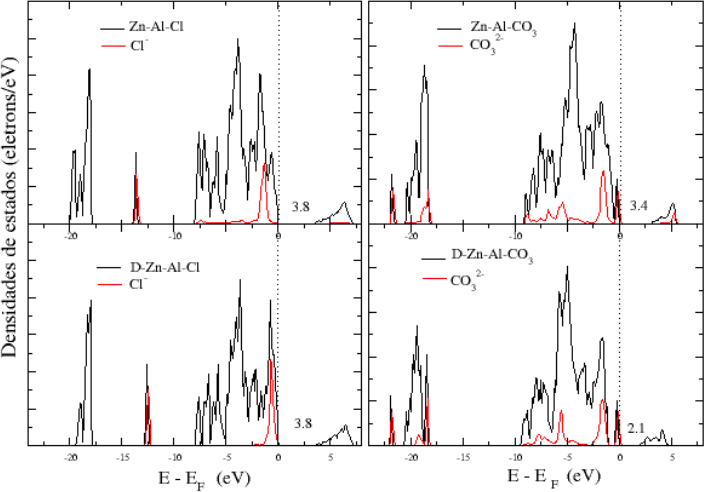
2 – Diferença de Densidade de Carga
As diferenças de densidade de carga nos informa a transferência de carga de uma parte do sistema com outra. Com a densidade total já conhecida, as densidades de cada parte do sistema são computadas e simplesmente subtraídas gerando, assim, plots com lóbulos de aumento de carga e de deficit de carga.
{kind=link}
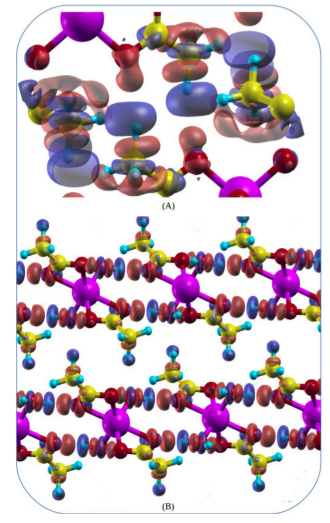
3 – Cargas de Bader
As cargas de bader são obtidas pela estimativa dos volumes de cada átomo do sistema pelas superfícies de fluxo zero. Essa análise é muito útil para a verificação de processos de fisissorção e quimissorção.
4 – Orbitais HOMO e LUMO
A visualização dos orbitais HOMO (Highest Occupied Molecular Orbital) e LUMO (Lowest Unoccupied Molecular Orbital) nos permite avaliar a estabilização de uma determinada espécie química adsorvida e auxiliar a interpretação das informações de basicidade/acidez obtidas pelos cálculos de DOS e pDOS.
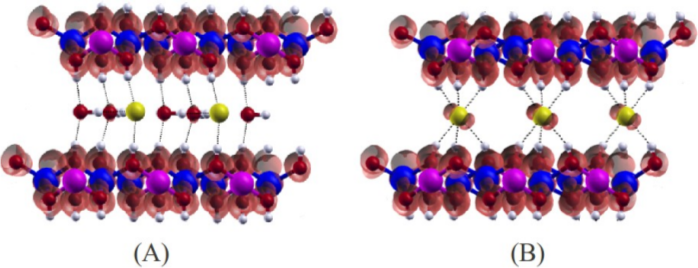
-
Simulação de Difratogramas
Depois de feita a otimização de geometria, vale a pena fazer a comparação dos difratogramas simulados e experimentais. Essa simulação é feita por cálculos de fator de estrutura e pela lei de Bragg. Em muitos casos, uma redução da célula unitária cristalográfica para a célula primitiva deve ser feita para diminuir os custos computacionais. Essa comparação de difratogramas permite justamente verificar se a otimização da célula primitiva não altera significantemente a simetria do composto.
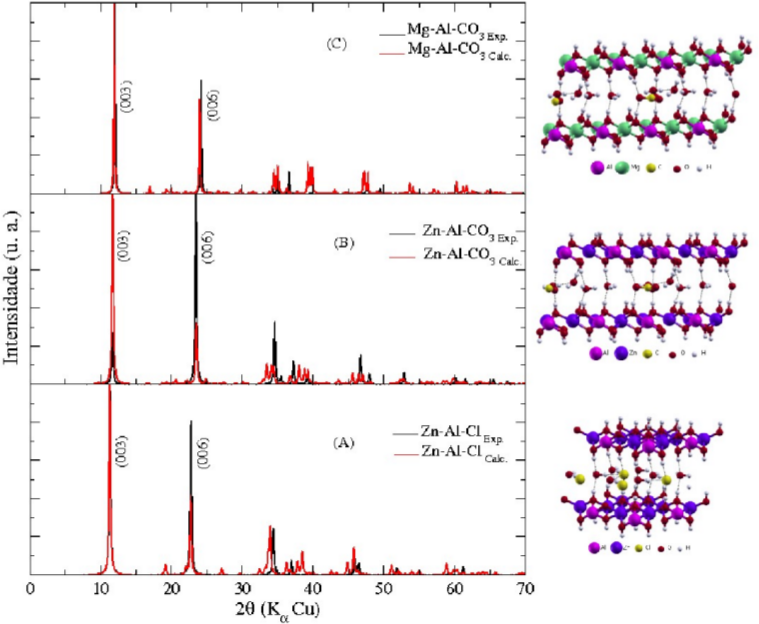
-
CI-NEB
Os cálculos de CI-NEB são feitos pela construção de imagens intermediárias entre duas imagens (reagente e produto) com a computação das forças reais e virtuais. Logo, o caminho de menor energia obtido (MEP – Minimum Energy Path) indica a forma como uma etapa de reação acontece e permite até estimar a sua energia de ativação.
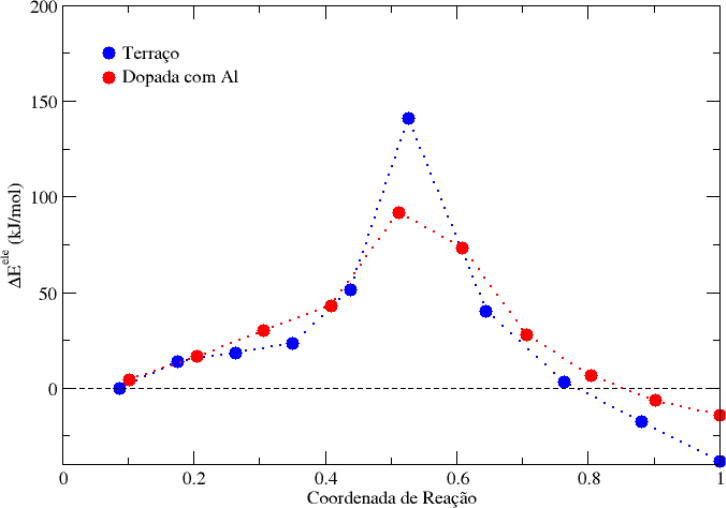
Referências:
TAVARES, S. R.; WYPYCH, F.; LEITÃO, A. A. A theoretical study of a homologous series of zinc n-alkanoates (2 ≤ nC ≤ 8): structural analysis, evaluation of their interactions and monofilm formation, Chemical Physics Letters, 636 (2015) 154-162
TAVARES, S. R.; VAISS, V. S.; WYPYCH, F.; LEITÃO, A. A. Theoretical study of the anion exchange properties and the thermal decomposition of Zn5(OH8)(NO3)2.2H2O and Zn5(OH)8(NO3)2.2NH3 Applied Clay Science 114 (2015) 103-111