
– O conjunto de práticas descrito nesta seção tem por objetivo formar usuários independentes do pacote Quantum-ESPRESSO https://www.quantum-espresso.org/
– Todas as práticas foram construídas a partir de tutoriais do Q-E recolhidos nas páginas dos grupos mantedores do pacote.
– Os programas PWgui e XCrysDen também são necessários para se realizar as práticas.
– É importante ressaltar que as práticas foram realizadas utilizando o pacote Quantum ESPRESSO v6.8, instalado no Linux Mint 20.3 Cinnamon.
Procedimentos Iniciais
Instalando o quantum-ESPRESSO:
Primeiro instale o MPICH e as bibliotecas matemáticas blas, lapack e fftw3-3:
~$ sudo apt install mpi-default-dev libopenmpi-dev libblas-dev liblapack-dev libfftw3-3
em seguida baixe, configure, compile e instale o quantum-espresso com os comandos:
~$ cd /opt
~$ sudo wget https://github.com/QEF/q-e/releases/download/qe-6.8/qe-6.8-ReleasePack.tgz
~$ sudo tar -vzxf qe-6.8-ReleasePack.tgz && sudo rm qe-6.8-ReleasePack.tgz
~$ cd qe-6.8
~$ sudo ./configure
~$ make all -j$(nproc)
~$ sudo make install
Instalando o xcrysden
Abra um terminal e dê um comando:
~$ sudo apt install xcrysden
Instalando o pwgui
O programa pwgui encontra-se compilado, de modo que para usá-lo basta fazer o download acessando a página do desenvolvedor, descompactar e executar.
Obs: Para poder usá-lo através do terminal, em qualquer diretório, basta movê-lo para: /usr/bin. Isso pode ser feito abrindo um terminal dentro da pasta onde ele foi descompactado e digitando o seguinte comando:
~$ sudo mv pwgui /usr/bin/
Iniciando as práticas
As práticas implementas são as seguintes:
– 1) Primeiros passos em semicondutores e isolantes:
– 2) Estrutura de bandas do Si:
– 3) Primeiros passos em metais:
– 4) Densidade de carga:
– 5) Análise de Bader:
– 6) Função de onda quadrática – Avaliação de orbitais:
– 7) Densidade de estados (DOS):
– 8) Fônons em Γ:
– 9) Superfícies:
– 10) Estado de transição:
– 11) Pseudopotenciais:
1. Primeiros passos em semicondutores e isolantes
A) Inicialmente, deve-se criar um diretório praticas onde as simulações serão desenvolvidas.
B) Agora, baixe o arquivo praticas-gfqsi.tar.gz (Se o download falhar, copie e cole o link em uma nova aba do navegador e dê enter) e descompacte-o dentro da pasta criada. É importante notar que todas as vezes que a pasta criada com o procedimento de descompactação for citada daqui pra frente no texto ela será referida como “/praticas-gfqsi/”, porém o caminho completo desse diretório vai variar de acordo com cada usuário e esse deve fazer as alterções necessárias.
Nessa prática são apresentados os programas PWgui, XCysDen e variáveis básicas do programa PWscf.
Testes iniciais com o Si
Alguns dados importantes sobre o Si: Simetria = FCC; parâmetro de rede = 5.43 Angstrom (1 Angstrom = 1.8897265 Bohr)
Diretório original: /praticas-gfqsi/semicondutores
1) Crie o diretório semicondutores dentro do seu diretório praticas. Copie os arquivos *.UPF do diretório original para o diretório semicondutores.
2) Dentro do pwgui, crie um arquivo para rodar um cálculo scf de Si.
2.1) executar dentro do diretório semicondutores
prompt> pwgui &
2.2) No menu principal selecione “Create new input file” e depois “Create new PW.X input file”
Na aba “Control” definir as variáveis:
title: 'Modelo inicial - testes inicias'
calculation: scf
outdir: diretório atual (use o botão "Directory ..." à direita)
wfcdir: diretório atual (use o botão "Directory ..." à direita)
pseudo_dir: diretório atual (use o botão "Directory ..." à direita)
prefix: 'si-modelo'
Na aba System:
ibrav: Free Lattice
celldm(1): 1.8897265
nat: 2
ntyp: 1
ecutwfc: 20
Na aba Lattice & Atomic data:
Enter Lattice Basis Vectors
0.0 2.715 2.715
2.715 0.0 2.715
2.715 2.715 0.0
Na seção Enter Atomic Types:
Defina Si em Atomic-label Defina a massa do silício como 0.0 por enquanto. Use o botão "Pseudopotential ..." para escolher o arquivo '''.UPF''' com o pseudopotencial para este elemento.
Em Atomic Coordinate Unit:
Selecione: (crystal)
Defina em Atomic Coordinates:
Si 0.0 0.0 0.0
Si 0.25 0.25 0.25
Na aba K-point data, selecionar:
(automatic) nk1: 8 nk2: 8 nk3: 8 sk1: 1 sk2: 1 sk3: 1
Selecione no menu principal o botão “Save the current input”, escreva como nome “si-modelo.pw.inp” e pressione o botão “Save” e depois “Yes”.
Energia de corte
Para estudar qual a melhor energia de corte para o sistema, deve-se fazer o cálculo de estrutura eletrônica com diferentes valores de ecut. O valor de ecut que será usado deve ser o menor valor no qual a energia total do sistema tenha convergido. Para realizar esta análise, deve criar um arquivo .dat com duas colunas, uma com os valores de ecut e outra com os valores de energia total e ao analisá-las escolhemos o menor valor de ecut que apresente o respectivo valor de energia total diferindo de um milésimo de Ry com o valor subsequente.
Para obter os valores de Energia de Corte de um sistema, os seguintes passos devem ser seguidos:
– Dentro do diretório semicondutores crie outro diretório teste-ecut. Para criar o arquivo sh, executar dentro do diretório teste-ecut:
prompt> vim silicio-ecut.sh
– O teste de convergência da energia de corte pode ser realizado colando o script abaixo:
#! /bin/sh
#Variaveis de ambiente
export OMP_NUM_THREADS=1
export CMD_PW="/usr/bin/pw.x"
export CALC_DIR="/home/<seu usuário>/<seu diretório>"
for ecut in 10 20 30 40 50 60 70 80;
do
cat>si-modelo-ecut-$ecut.pw.inp<<EOF
&CONTROL
title = 'Silicio ecut $ecut' ,
calculation = 'scf' ,
outdir = './' ,
wfcdir = './' ,
pseudo_dir = './' ,
prefix = 'si-modelo-ecut-$ecut' ,
/
&SYSTEM
ibrav = 0 ,
celldm(1) = 1.8897265 ,
nat = 2 ,
ntyp = 1 ,
ecutwfc = $ecut ,
ecutrho = $(($ecut * 8)) ,
/
&ELECTRONS
/
CELL_PARAMETERS cubic
0.000000000 2.715000000 2.715000000
2.715000000 0.000000000 2.715000000
2.715000000 2.715000000 0.000000000
ATOMIC_SPECIES
Si 28.0855 Si.pw91-n-van.UPF
ATOMIC_POSITIONS crystal
Si 0.000000000 0.000000000 0.000000000
Si 0.250000000 0.250000000 0.250000000
K_POINTS automatic
8 8 8 1 1 1
EOF
$CMD_PW < $CALC_DIR/si-modelo-ecut-$ecut.pw.inp > $CALC_DIR/si-modelo-ecut-$ecut.pw.out
done
– Observação: Caso haja erro no cálculo, ir no arquivo silicio-ecut.sh e deletar o espaço na frente de EOF.
– Note no script que você deve definir o diretório no qual o teste será realizado. Execute o arquivo silicio-ecut.sh:
prompt> sh silicio-ecut.sh &
– Para verificar as energias basta executar o seguinte comando:
prompt> grep ! si-modelo-ecut-*.pw.out
– Selecione os dados obtidos e execute o seguinte comando:
prompt> vim si-teste-ecut.dat
O arquivo deve conter duas colunas: energia total (Ry) x ecut
Utilize os valores da energia eletrônica total e os da energia de corte em cada um para reproduzir um gráfico que lhe fornece o valor da menor energia de corte, semelhante ao mostrado nesta figura.
Amostragem de pontos-k
O procedimento para a escolha dos pontos k é análogo ao procedimento de escolha da energia de corte, porém alterando a variável k-points na aba de mesmo nome em x, y e z e também no ponto gamma.
Dentro do diretório semicondutores crie outro diretório denominado de teste-kpts. Após a análise de convergência do teste ecut, escolha o valor adequado para a energia de corte, crie um novo arquivo sh (silicio-kpts.sh) e copie o script acima para realizar o teste de convergência para a amostragem de pontos k. No novo arquivo sh, altere os diretórios para a nova pasta e substitua as seguintes linhas:
for ecut in 10 20 30 40 50 60 70 80; do
cat>si-modelo-ecut-$ecut.pw.inp<<EOF
title = 'Silicio ecut $ecut' ,
prefix = 'si-modelo-ecut-$ecut' ,
ecutwfc = $ecut ,
8 8 8 1 1 1
$CMD_PW < $CALC_DIR/si-modelo-ecut-$ecut.pw.inp > $CALC_DIR/si-modelo-ecut-$ecut.pw.out
Por:
for kpts in 1 2 3 4 5 6 7 8 9 10; do cat>si-modelo-kpts-$kpts.pw.inp<<EOF title = 'Silicio kpts $kpts' , prefix = 'si-modelo-kpts-$kpts' , ecutwfc = <o valor de ecut em que a energia total converge>, ecutrho = <(o valor de ecut em que a energia total converge)*8> $kpts $kpts $kpts 1 1 1 $CMD_PW < $CALC_DIR/si-modelo-kpts-$kpts.pw.inp > $CALC_DIR/si-modelo-kpts-$kpts.pw.out
Repita o mesmo procedimento para a análise do resultado do teste de convergência da energia de corte e construa o gráfico da convergência da energia eletrônica com a amostragem de pontos k, semelhante ao mostrado aqui.
- Chadi, D. J., Cohen, M. L., Physical Review B volume 8, number 12 (1973)
- Monkhorst, H. J., Pack, J. D., Physical Review B volume 13, number 12 (1976)
- Moreno, J., Soler, J. M., Physical Review B volume 45, number 24 (1992)
Amostragem de pontos-gamma
Calcule também o ponto gamma para comparar com os outros valores de amostragem. Execute o comando:
cp si-modelo-kpts-$kpts.pw.inp si-modelo-kpts-gamma.pw.inp
Abra o PWgui:
prompt> pwgui &
No menu principal selecione Open an existing input file e depois Open an existing PW.X input file e selecione o arquivo criado si-modelo-kpts-gamma.pw.inp:
Na aba K-point data, selecionar:
(gamma)
Selecione no menu principal o botão Save the current input, depois Yes. Execute o arquivo si-modelo-kpts-gamma.pw.inp.
prompt> pw.x < si-modelo-kpts-gamma.pw.inp > si-modelo-kpts-gamma.pw.out &
Otimização da geometria dos átomos no Si
Execute o pwgui dentro do seu diretório semicondutores
prompt> pwgui &
Abra o arquivo si-modelo.pw.inp usando o botão Open an existing input e o botão Open an existing PW.X file
Na aba Control, troque calculations de scf para relax e em prefix mudar para ‘si-opt’
Na aba Lattice & Atomic data adicionar 1 1 1 após as coordenadas dos átomos (permitirá a otimização de posição)
Salve o arquivo com o nome si-opt.pw.inp usando o o botão Save the current input under different name
Examine o arquivo de entrada e execute a simulação
prompt> vim si-opt.pw.inp prompt> pw.x < si-opt.pw.inp > si-opt.pw.out &
Verifique o arquivo de saída
prompt> vim si-opt.pw.out
Otimização completa da geometria do Si
Execute o pwgui dentro do seu diretório semicondutores
prompt> pwgui &
Abra o arquivo si-modelo.pw.inp usando o botão Open an existing input e o botão Open an existing PW.X file
Na aba Control, troque calculations de scf para vc-relax e em prefix mudar para ‘si-fullopt’
Na aba Lattice & Atomic data adicionar 1 1 1 após as coordenadas dos átomos (permitirá a otimização de posição)
Na aba Variable Cell escreva:
cell_dynamics = damp-w , wmass = 0.75 x massa total/ (π**2) , (para Wentzcovitch)
OBS: Calcule o valor do wmass utilizando a soma das massas de todos os átomos da célula unitária. Para isso , nesse caso a massa de cada átomo de Si = 28.09 e para todos os casos π = 3.14159265.
Salve o arquivo com o nome si-fullopt.pw.inp usando o o botão Save the current input under different name
Examine o arquivo de entrada e execute a simulação
prompt> vim si-fullopt.pw.inp
prompt> pw.x < si-fullopt.pw.inp > si-fullopt.pw.out &
Verifique o arquivo de saída
prompt> vim si-fullopt.pw.out
2. Estrutura de bandas do Si (voltar ao índice)
Diretório original: /praticas-gfqsi/Bandas
1) Crie um diretório “bandas” dentro do seu diretório “praticas”. A partir do diretório original copie os seguintes arquivos para o diretório “bandas”:
1.1) si.scf.in: arquivo pronto para o cálculo scf no Si. (/praticas-gfqsi/Bandas/)
1.2) Si.pw91-n-van.UPF: pseudopotencial para Si. (/praticas-gfqsi/semicondutores/)
1.3) si-bands.in: arquivo de entrada o programa bands.x (/praticas-gfqsi/Bandas/)
1.4) INPUT_BANDS: instruções para montagem do arquivo de entrada para o programa bands.x. (/praticas-gfqsi/Bandas)
2) Cálculo scf
2.1) Examinar o arquivo de entrada
prompt> vim si.scf.in
OBS: Fazer as devidas modificações em outdir, wfcdir e pseudo_dir.
2.2) Executar os comandos:
prompt> mv si.scf.in si.scf.inp
prompt> pw.x <si.scf.inp> si.scf.out
prompt> vim si.scf.out
3) Cálculo de estrutura de bandas (nscf)
3.1) Abra o Xcrysden (caso já não esteja aberto)
prompt> xcrysden &
Uma vez no XcrysDen, abra o arquivo si.scf.inp e vá em Tools no menu principal e em seguida clique em k-path Selection.
Feito isso, será aberta um janela com a figura da primeira zona de Brillouin explicitando os pontos especiais. Escolha a seguinte seqüência de pontos:
L (centro do hexagono),
Γ (centro da figura),
X (centro do quadrado),
W (vértice desse quadrado),
Γ novamente.
Clique em OK. Selecione Total number of k-points along the path como 36 e clique em OK. Selecione e escreva si-kpt.pwscf em File name e clique em Save.
3.2) Examine os novos arquivos criados
prompt> vim supportInfo.kpath
prompt> vim si-kpt.pwscf
3.3) Crie o arquivo si.nscf.inp
prompt> cat si.scf.inp si-kpt.pwscf > si.nscf.inp
3.4) Edite o novo arquivo:
prompt> vim si.nscf.inp
Uma vez dentro do arquivo si.nscf.inp, apague as linhas:
K_POINTS automatic 8 8 8 1 1 1
Acrescente dentro da namelist &system a variável nbnd = 8, para que sejam calculadas 8 bandas. Os comandos devem sempre estar separados por vírgulas e deve haver uma vírgula depois do último comando em cada nemelist. Em &control, na variável calculation troque scf por nscf. Grave de novo o arquivo si.nscf.in. Observação Importante: “prefix” tem que ser o mesmo do cálculo scf, porque a densidade e potencial deste serão usadas no cálculo nscf.
3.5) Faça o cálculo da estrutura de bandas
prompt> pw.x < si.nscf.in > si.nscf.out &
3.6) Examine o arquivo de sáida:
prompt> vim si.nscf.out
Tente responder as seguintes questões: Quantas iterações foram feitas? Que potencial foi usado na diagonalização? Que wfcs foram usadas como chute inicial (inital guess)? A densidade de carga e a energia total foram computadas?
3.7) Gere o arquivo .out contendo apenas as bandas:
prompt> vim si-bands.in
prompt> bands.x < si-bands.inp > si-bands.out
OBS: Mude o arquivo si-bands.in para a extensão .inp e fazer as devidas modificações em outdir e filband.
3.8) Verifique o arquivo si.nscf.bands
prompt> vim si.nscf.bands
4) Gráfico de estrutura de bandas
prompt> plotband.x
Input file > si.nscf.bands Emin, Emax > -10, 20 output file (gnuplot/xmgr) > xmgr output file (ps) > si.bands.ps Efermi > < valor de 'highest occupied level (ev)' encontrado no .out do cálculo scf> deltaE, reference E (for tics) > 5, <Efermi>
visualize o arquivo si.bands.ps e compare com este si.bands.png
3. Primeiros passos em metais (voltar ao índice)
Aqui são apresentadas variáveis básicas de ocupação de estados que são necessárias ao cálculo de estrutura eletrônica de sistemas metálicos.
Testes iniciais com o Al
Alguns dados importantes sobre Al: Simetria = FCC; Parâmetro de rede = 4.05 Angstrom (1 Angstrom = 1.8897265 Bohr)
Diretório original: /praticas-gfqsi/metais
1) Crie o diretório metais dentro do seu diretório praticas. Copie os arquivos *.UPF do diretório original para o diretório metais.
2) Dentro do pwgui, crie um arquivo para rodar um cálculo scf de Al.
2.1) executar dentro do diretório metais
prompt> pwgui &
2.2) No menu principal selecione “Create new input file” e depois “Create new PW.X input file”
Na aba Control definir as variáveis:
title: 'Modelo inicial - testes inicias'
calculation: scf
outdir: diretório atual (use o botão "Directory ..." à direita)
wfcdir: diretório atual (use o botão "Directory ..." à direita)
pseudo_dir: diretório atual (use o botão "Directory ..." à direita)
prefix: 'al-modelo'
Na aba System:
Na seção Required variables :
ibrav: Free Lattice
celldm(1): 1.8897265
nat: 1
ntype: 1
ecutwfc: 20
Na seção Optional variables
occupations: smearing
degauss: 0.03
smearing: gaussian
Na aba Lattice & Atomic data:
Enter Lattice Basis Vectors
0.0 2.025 2.025
2.025 0.0 2.025
2.025 2.025 0.0
Na seção Enter Atomic Types:
Defina Al em Atomic-label
Defina a massa da Al como 0.0, por enquanto.
Use o botão "Pseudopotential ..." para escolher o arquivo '''.UPF'''com o pseudopotencial para este elemento.
Em Atomic Coordinate Unit:
Selecione: (crystal)
Defina em Atomic Coordinates:
Al 0.0 0.0 0.0
Na aba K-point data, selecionar: (automatic)
nk1: 8 nk2: 8 nk3: 8
sk1: 1 sk2: 1 sk3: 1
Selecione no menu principal o botão “Save the current input”, escreva como nome “al-modelo.pw.inp” e pressione o botão “Save” e depois “Yes”.
As variáveis definidas na seção Optional Variables da aba System estão relacionadas à natureza metálica do sistema. Em metais, ocorre cruzamento entre as bandas de valência (BV) e condução (BC). Com isso, há uma probabilidade não-nula de ocupação de estados da BC mesmo a temperatura T=0K. Dessa forma, em sistemas metálicos, a distribuição dos elétrons nas bandas deve ser feita através de uma “função de smearing” (daí a opção ‘smearing’ na variável occupations), que pode assumir diversas formas. O PWscf oferece várias possibilidades para fazer essa distribuição, de acordo com a variável smearing. Todas elas acham correspondentes (e breves resumos) na definição da variável occopt do ABINIT (http://www.abinit.org/Infos_v5.5/input_variables/varbas.html#occopt).
Energia de corte
Realizar o mesmo procedimento descrito para o silício, com os seguintes valores de energia de corte 10 20 30 40 50 60 70 80.
Amostragem de pontos-k
Repetir o procedimento descrito na prática de semicontutores, variando os valores de kpts de 1 a 10.
4. Densidade de carga (voltar ao índice)
Obtenção do densidade de carga no plano 110 do Si (Obs: plano 110 da célula primitiva).
Diretório original: /praticas-gfqsi/Bandas
1) A obtenção da densidade de carga será feita a partir do cálculo SCF da prática anterior “obtenção da estrutura de bandas do Si”. Assim sendo, deverá ser usado o mesmo diretório.
2) Preparação do arquivo de entrada para pp.x
prompt> pwgui &
Uma vez no pwgui, abra um novo arquivo de entrada para pp.x e especifique as variáveis:
Na aba Specify property to calculate:
prefix: ‘silicon’
outdir: “selecione o diretório onde estão os arquivos”
filplot: ‘sicharge’
plot_num “charge density”
Na aba Specify Plot:
nfile: 1
filepp(1): ‘sicharge’
weight(1): 1.0
fileout: ‘si.rho.dat’
iflag: “2D plot”
output_format: “format suitable for plotrho”
e1(1): 1 e1(2): 1 e1(3): 0
e2(1): 0 e2(2): 0 e2(3): 1
nx: 56 ny: 40
Gravar o arquivo como si-rho.inp
3) Cálculo da densidade de carga no plano 110
prompt> pp.x < si-rho.inp > si-rho.out
4) Gerando a figura:
prompt> plotrho.x
O programa rodará interativamente:
Selecione entrada como si.rho.dat (enter).
Selecione o nome que preferir para o arquivo output si-rho.ps (enter)
O programa perguntará: Logarithmic scale (y/n)? responda: n (enter)
Para as váriáveis: min, max, # of levels; escreva, por exemplo 0.01 0.1 6 (enter)
Visualize o arquivo si.rho.ps. Deve-se obter um resultado semelhante a este si.rho.png
Rode de novo o plotrho.x e troque as váriáveis: min, max, # of levels. Observe que min e max devem estar dentro do intervalo “Bandas”, apresentado na linha acima e que “# of levels” dará o número de faixas de cores da figura. Verifique suas novas figuras.
Densidade de carga 3D
Diretório original: /praticas-gfqsi/carga3D
Assim como é possível gerar a densidade de carga em um plano cristalográfico qualquer, também é possível obtê-la em três dimensões. Este resultado é particularmente útil para a análise da densidade de carga ligante (próxima prática).
Para gerar a densidade de carga em 3D, siga o seguinte procedimento:
– Crie um diretório chamado cargas3D dentro da pasta de práticas e copie os arquivos *.scf.inp e *.UPF do Diretório original para ele.
OBS: a partir daqui, o arquivo agua.scf.inp será usado como exemplo. (O passo-a-passo deve ser realizado também para os arquivos lamela.scf.inp e tudo.scf.inp)
– Edite os campos outdir e wfcdir para o diretório cargas3D.
– Rode o cálculo scf acima com o pw.x
– A seguir, use o PWGUI para criar um arquivo de entrada para o pp.x com as seguintes especificações:
Na aba Specify property to calculate:
prefix: agua.pw (o mesmo que foi utilizado no cálculo com o pw.x para a estrutura)
outdir: “o diretório que contém o .save da estrutura”
filplot: agua_charge
plot_num: charge density
Na aba Specify Plot:
nfile: 1
filepp (1): (default: filepp = filplot)
weight (1): 1.0
fileout: agua.charge.xsf
iflag: 3D plot
output_format: XCRYSDEN’S XSF format (whole unit cell)
Salve o arquivo com o nome de agua.charge.pp.inp e rode o pp.x:
prompt> pp.x < agua.charge.pp.inp > agua.charge.pp.out
Visualize o arquivo .xsf gerado com o XCrySDen:
prompt> xcysden &
– Na aba File clique em Open Structure depois em Open XSF (XCrySDen Structure File) selecione o arquivo agua.charge.xsf
– Clique em translational asymmetric unit e depois em orient to XZ plane
– Na aba “Tools”, selecione “Data grid” e dê “ok”. Um bom valor para isovalue é 0.003, marque a opção Render +/- isovalue e clique em submit.
– Deve-se obter uma figura parecida com esta aqui agua.charge.png
– Repita o procedimento para os arquivos lamela.scf.inp e tudo.scf.inp, salvando todos os passos no mesmo diretório cargas3D. Observe no Xcrysden .xsf de cada material e compare as respectivas densidades de cargas.
Densidade de carga ligante
Quando se trabalha com adsorção de moléculas em superfícies, nem sempre é fácil afirmar se estamos lidando com um caso de fisissorção (apenas interação eletrostática) ou quimissorção (ligação química). Uma ferramenta que possibilita esse tipo de análise é a densidade de carga ligante, que nada mais é que a diferença de densidade de carga entre o sistema “superfície + adsorvato” e suas partes isoladas.
Para fazer esse cálculo, é preciso, primeiro, gerar a densidade de carga 3D do sistema “superfície + adsorvato”, da “superfície” isolada e do “adsorvato” isolado. Utilizando os exemplos da prática anterior, seriam, respectivamente, os arquivos tudo_charge, lamela_charge e agua_charge.
A seguir, basta construir um novo arquivo de entrada para o pp.x (ou editá-lo a partir do arquivo de entrada para o cálculo da densidade de carga do sistema completo):
Na aba Specify property to calculate:
prefix: tudo.pw
outdir: “o diretório do .save”
filplot: tudo_chargeDIFF
plot_num: charge density
Na aba Specify Plot:
nfile: 3 (isso abrirá mais dois campos de filepp)
filepp (1): tudo_charge(é recomendável que o primeiro seja o do sistema completo)
filepp (2): lamela_charge filepp (3): agua_charge
Obs1: todos esses arquivos *_charge são gerados durante a obtenção da densidadede carga 3D.
weight (1): 1.0
weight (2): -1.0
weight (3): -1.0
Obs2: os pesos negativos são porque essas densidades serão subtraídas da densidade do sistema completo.
fileout: tudo.chargeDIFF.xsf
iflag: 3D plot
output_format: XCRYSDEN’S XSF format (whole unit cell)
Salve o arquivo com o nome de tudo.chargeDIFF.pp.inp e rode o pp.x:
prompt> pp.x < tudo.chargeDIFF.pp.inp > tudo.chargeDIFF.pp.out
Visualize o arquivo .xsf gerado com o XCrySDen:
prompt> xcysden &
– Na aba File clique em Open Structure depois em Open XSF (XCrySDen Structure File) selecione o arquivo tudo.chargeDIFF.xsf
– Clique em translational asymmetric unit e depois em orient to XZ plane
– Na aba “Tools”, selecione “Data grid” e dê “ok”. Um bom valor para isovalue é 0.003, marque a opção Render +/- isovalue e clique em submit.
– Deve-se obter uma figura parecida com esta aqui tudo.chargeDIFF.png
O código de cores “default” do XCrySDen indica com a cor vermelha as regiões onde a densidade de carga do sistema completo aumentou e com a cor azul as regiões onde a ela diminuiu. Ou seja, esse resultado mostra a migração da densidade de carga pelo sistema. Migrações de densidade no sentido de formação de novas ligações já apreciáveis com “isovalue” 0.003, em geral, podem ser consideradas como evidência de quimissorção. Fisissorções só vão apresentar migrações de carga apreciáveis em valores de “isovalue” da ordem de décimos de milésimos.
5. Análise de Bader (voltar ao índice)
Para o cálculo da análise de Bader, deve-se rodar o programa pp.x. O arquivo poder ser o mesmo arquivo usado para o cálculo da densidade de cargas. (Prática anterior)
1) Digite no prompt:
$ cp arquivo-da-densidade-de-cargas.pp.inp arquivo-da-analise-de-Bader.pp.inp
Usualmente, os arquivos utilizados nos cálculos de densidade de cargas possuem a variável output_format com o valor atribuído a 5. Isso significa que o cálculo irá gerar um arquivo para ser visualizado no XCrysDen. Para o cálculo da análise de Bader esse valor deve ser 6, que gera um arquivo de formato Gaussian (cube-file). Portanto, deve-se modificar o valor da variável output_format para 6 e na variável fileout escrever: nomedoarquivo.cube.
2) Em seguida realizar o cálculo:
prompt> pp.x < arquivo.Bader.pp.inp > arquivo.Bader.pp.out &
Rodar o programa bader (Se o download falhar, copie e cole o link em uma nova aba do navegador e dê enter) com o arquivo .cube gerado. (para saber mais sobre este software visite: http://theory.cm.utexas.edu/henkelman/code/bader/)
No terminal digite:
$ bader arquivo.cube
Os valores das cargas para cada átomo é escrito no arquivo ACF.dat
Para descontar os valores de valência desse resultado utiliza-se o programa badervalencia (Se o download falhar, copie e cole o link em uma nova aba do navegador e dê enter).
Esse programa tem como função descontar a valência da carga total em cada átomo fornecida pelo programa bader. Após utilização dele, o resultado é a carga de cada átomo em um dado sistema.
O programa badervalencia é simples e deve ser usado da seguinte forma:
$ python3 badervalencia.py
O programa vai pedir o arquivo de saída do cálculo scf e depois o arquivo ACF.dat (gerado pelo programa bader). Por fim, ele vai pedir um nome para salvar os dados.
6. Função de onda quadrática – Avaliação de orbitais (voltar ao índice)
Diretório original: /praticas-gfqsi/Orbitais
Para avaliar orbitais, tais como:
– Orbitais HOMO ou LUMO de uma molécula,
– Orbitais que contribuem majoritariamente para a banda de valência ou para a banda de condução num sólido,
deve-se preparar um cálculo do quadrado da densidade de carga. Este é um cálculo de pós-tratamento, por isto, deve-se utilizar uma geometria já otimizada, para posteriormente adotar os passos mostrados a seguir.
Para exemplificar serão apresentados os procedimentos para se obter os orbitais HOMO e LUMO da molécula de água.
1) Agora, baixe o arquivo Orbitais (Se o download falhar, copie e cole o link em uma nova aba do navegador e dê enter) e descompacte-o dentro do seu diretório de praticas.
2) Edite os arquivos, direcionando os arquivos temporários para o seu endereço.
3) Faça um cálculo “scf” utilizando a geometria já otimizada.
prompt> pw.x <agua.scf.inp> agua.scf.out &
4) Visualize o arquivo de saída.
prompt> vim agua.scf.out
Observe que no início do arquivo é apresentado o número de bandas ocupadas,
number of Kohn-Sham states= 4
Seja-se avaliar o orbital HOMO da molécula, então, deve-se investigar a 4° banda (a banda ocupada de maior energia). Além disso, deve-se incluir alguns orbitais virtuais para a avaliação do LUMO. Para isto, deve-se fazer um cálculo “nscf”.
5) Visualize o arquivo de entrada
prompt> vim agua.nscf.inp
Observe que a diferença deste arquivo para o anterior está na inclusão da variável nbnd = 6. Esta variável informa quantas bandas serão consideradas no cálculo “nscf”. Aqui, optou-se por acrescentar duas bandas virtuais, mas só a 5° (orbital desocupado de menor energia) será realmente investigada.
6) Faça o cálculo “nscf”
prompt> pw.x <agua.nscf.inp> agua.nscf.out &
Para gerar o quadrado das densidades de carga, deve-se cálcular os arquivos de pós tratamento “agua-homo.pp.inp” e “agua-homo.pp.inp”. Antes, visualize estes arquivos.
prompt> vim agua-homo.pp.inp
Ao invés de ver o arquivo pelo terminal é possível abri-lo no “pwgui” para ter mais detalhes sobre as variáveis, neste caso, faça
prompt> pwgui &
Vá em file – Open – Open PP.X input – “agua-homo.pp.inp”
Atente para as seguintes variáveis: “prefix”, “plot_num”, “kpoint”, “kband” e “lsign”.
prefix – deve ser igual ao dos arquivos anteriores (Ex. todos os arquivos têm “prefix = agua”)
kpoint – deve indicar em que ponto k o quadrado da densidade de carga será calculado. Os pontos k são númerados (ver no arquivo de saída do cálculo “nscf”).
number of k points= 1
cart. coord. in units 2pi/a_0
k( 1) = ( 0.0000000 0.0000000 0.0000000), wk = 2.0000000
Aqui há somente um ponto k, pois o cálculo foi realizado no “ponto Gama”.
kband – deve indicar qual banda será investigada. Aqui, por motivos já discutidos anteriormente, será escolhido a 4° e 5° banda (a banda ocupada de maior energia e a banda desocupada de menor energia, respectivamente).
lsign – só pode ser incluída quando o cálculo é realizado no ponto Gama.
7) Rodar os arquivos de pós-processamento.
prompt> pp.x <agua-homo.pp.inp> agua-homo.pp.out & prompt> pp.x <agua-lumo.pp.inp > agua-lumo.pp.out &
8) Visualizar as imagens do quadrado da densidade de carga.
prompt> xcrysden &
File – Open structure – Open XSF – “agua-homo.xsf” (depois, proceda de maneira analoga com o arquivo “agua-lumo.xsf”) Na aba “Tools”, selecione “Data grid” e dê “ok”. Selecione a opção Render +/- isovalue. Um bom valor para isovalue é 0.03 para o “agua-homo.xsf” e 0.003 para o “agua-lumo.xsf”.
O código de cores “default” do XCrySDen indica com a cor vermelha as regiões onde a densidade de carga do sistema aumentou e com a cor azul as regiões onde a ela diminuiu. Ou seja, esse resultado mostra a migração da densidade de carga pelo sistema.
7. Densidade de estados (DOS) (voltar ao índice)
Densidade de Estados no Ni.
Diretório original: /praticas-gfqsi/DOS
1) Crie um diretório DOS dentro do seu diretório praticas. A partir do diretório original copie os seguintes arquivos para o diretório DOS:
prompt> mkdir DOS && cd DOS
prompt> cp /praticas-gfqsi/DOS/* .
2) Visualize e execute o arquivo ni.scf.inp:
prompt> vim ni.scf.inp
prompt> pw.x <ni.scf.inp> ni.scf.out
3) Visualise os arquivos criados:
prompt> vim ni.scf.out
4) Efeito da magnetização.
4.1) Copie o arquivo de saída do cálculo:
prompt> cp ni.scf.out ni.scf-0mag.out
4.2) Edite o arquivo scf:
prompt> vim ni.scf.inp
Em &System, acrescente as variáveis starting_magnetization(1)=0.7 e nspin=2.
4.3) Rode novamente o shell-script:
prompt> pw.x <ni.scf.inp> ni.scf.out
Compare os arquivos ni.scf-0mag.out e ni.scf.out e, verifique as diferenças na energia e na magnetização obtidas ao final do cálculo. O que significa magnetização? O que significa magnetização dentro do PWscf?
5) Cálculo não-scf. Vizualize o arquivo ni.nscf.inp:
prompt> vim ni.nscf.inp
Observe os valores e labels para as variáveis calculation, nbnd, starting_magnetization(1) e K_POINTS.
prompt> ./run-ni.nscf &
6) Obtenção da densidade de estados:
6.1) Observe a variável fildos e depois faça o cálculo:
prompt> dos.x <ni.dos.inp> ni.dos.out
6.2) Visualize os arquivos:
prompt> vim ni.dos.inp
prompt> vim ni.dos.out
prompt> vim ni.dos
7) Preparando e visualisando as figuras:
As figuras serão preparadas utilizando o xmgrace.
prompt> xmgrace &
Na aba Data, clicar em Import e depois em ASCII procurar o arquivo gerado ni.dos. e visualise o gráfico. que deve ter um aspecto similar ao mostrado nesta figura
7.1 Projeção da Densidade de Estados (PDOS) (voltar ao índice)
A Projeção da Densidade de Estados é um cálculo que permite tratar individualmente a Densidade de Estados de cada elemento de uma determinada estrutura. Logo, os cálculos são semelhantes e os diretórios devem ser os mesmos.
1) Crie um arquivo de entrada para o ProjWFC.x dentro do diretório DOS:
prompt> pwgui &
Clique no botão Create New Input File e depois selecione Create new ProjWFC.X input file. Então, construa seu arquivo de entrada.
2) Na aba Optional variables:
prefix = 'ni'
outdir = diretório DOS
filpdos = 'ni'
ngauss = Methfessel-Paxton of order 1
degauss = 0.02
DeltaE = 0.1
Emin = 5.0
Emax = 25.0
3) Modificar o nome do arquivo para ni.pdos.inp. Rode o processo:
prompt> projwfc.x <ni.pdos.inp> ni.pdos.out &
OBS: Os arquivos gerados possuem parênteses () no nome. Renomeie-os retirando os (), para que se possa visualiza-los com o comando vim. Exemplo: Renomeie-os para ni.pdos_atm#1Ni_wfc#1_s e ni.pdos_atm#1Ni_wfc#2_d.
4) Preparando e visualizando as figuras.
prompt> xmgrace &
Na aba Data, clicar em Import e depois em ASCII procurar os arquivos ni.pdos_atm#1Ni_wfc#1_s , ni.pdos_atm#1Ni_wfc#2_d e ni.pdos_tot, importe-os e observe a contribuição de de cada orbital no gráfico. Um exemplo pode ser visto aqui
8. Fônons em Γ (voltar ao índice)
Fônons em Γ para a brucita (hidróxido de magnésio).
Alguns dados importantes sobre a brucita:
– Simetria: hexagonal;
– Parâmetros de rede: a= b= c= 4.23 Angstroms (1 Angstrom = 1.8897265 Bohr).
Diretório original: /praticas-gfqsi/Fonons
1) Baixe o arquivo Fonons (Se o download falhar, copie e cole o link em uma nova aba do navegador e dê enter) e descompacte-o dentro do seu diretório de práticas.
2) Verifique o arquivo de entrada bru-scf.inp:
prompt> vim bru-scf.inp
Faça as devidas modificações em outdir, wfcdir e pseudo_dir abrindo o PWgui: correção na aba K-point data (no ponto &Gamma), definir crystal no campo K-Point input, o número de pontos-k (Number of K-Points) como 1 e, então, definir as coordenadas KX, KY e KZ abaixo como 0.0 0.0 0.0 e o peso (weight) como 1.0.
3) Executar o cálculo scf do arquivo bru-scf.inp:
prompt> pw.x <bru-scf.inp> bru-scf.out &
OBS: Em alguns sistemas a correção na aba K-point data pode ser feita fora do ponto gama. Neste caso definir automatic no campo K-Point input e, então, defina nk1, nk2, nk3 como 1 1 1 no campo K-Point mesh + grid (sk1, sk2 e sk3 devem permanecer como 1 1 1).
Neste momento você calculou a densidade eletrônica que será utilizada para o cálculo das frequências vibracionais.
4) Cálculo da matriz de forças. Prepare um arquivo de entrada para o programa ph.x:
prompt> pwgui &
4.1) Clique no botão Create new input file e depois em Create new input PH.X file:
jobtitle: 'Calculo de fonons do sistema brucita'
4.2) Na aba Files/Diretories:
outdir: selecionar o diretório usando o botão à direita
prefix: selecionar o mesmo prefix do cálculo scf - bru-t2
fildyn: bru.dyn.mat
4.3) Na aba What to Compute selecione:
trans: yes
epsil: yes
4.4) Na aba Control Options coloque:
recover: yes
ntyo: 3
amass (1): 24.00000
amass (2): 16.00000
amass (3): 1.00000
tr2_ph: 1.D-14
4.5) Na aba Suffix Cards coloque:
xq(1): 0 xq(2): 0 xq(3): 0
4.6) Gravar o arquivo como bru.ph.inp. Em seguida, verifique o arquivo criado e execute o cálculo ph.x:
prompt> vim bru.ph.inp
prompt> ph.x <bru.ph.inp> bru.ph.out &
4.7) Observe o arquivo de saída (bru.ph.out) e o arquivo contendo a matriz de forças (bru.dyn.mat):
prompt> vim bru.ph.out
prompt> vim bru.dyn.mat
4.8) Correção ASR e intensidades
Verifique o arquivo de entrada para o cálculo (bru.dyn.inp) e fazer as devidas modificações em fildyn e filmol:
prompt> vim bru.dyn.inp fildyn: o mesmo para do arquivo bru.ph.inp filmol: igual o fildyn, trocando bru.dyn.mat por bru.dyn.mol
Faça a simulação e examine o arquivo de saída (bru.dyn.out).
prompt> dynmat.x <bru.dyn.inp> bru.dyn.out
Obverve os modos vibracionais e as intensidades: as animações dos modos vibracionais podem ser visualizadas pelo programa molden abrindo o arquivo .mol ou as forças que correspondem a esses modos podem ser visualizadas pelo programa XCrysden usando o arquivo dynmat.axsf gerado.
Pelo molden: (para instalá-lo: prompt> sudo snap install molden)
prompt> vim bru.dyn.out prompt> molden bru.dyn.mol &
Selecione o botão Norm. modes no canto superior direito.
Verifique todos os modos vibracionais desejados (de rede e da molécula).
9. Superfícies (voltar ao índice)
Simulação da superfície (001) do Alumínio.
Diretório original: /praticas-gfqsi/superficies
1) Baixe o arquivo superficies (Se o download falhar, copie e cole o link em uma nova aba do navegador e dê enter) e descompacte-o dentro do seu diretório de práticas. Em seguida, copie o arquivo Al.pw91-n-van.UPF para esse diretório.
2) Cálculo scf.
2.1) Dentro do seu diretório superficies digite:
prompt> pwgui &
Dentro do PWgui, crie um arquivo para rodar um cálculo scf de Al.
2.2) Acessando o PWgui, clique no botão Create New Input File e depois selecione Create new PW.X input file. Então, construa seu arquivo de entrada.
2.2.1) Na aba Control defina as variáveis:
title: 'Al001-scf.pw'
calculation: scf
outdir: diretório atual (use o botão "Directory ..." à direita)
wfcdir: diretório atual (use o botão "Directory ..." à direita)
pseudo_dir: diretório atual (use o botão "Directory ..." à direita)
prefix: 'Al001-scf.pw'
2.2.2) Na aba System entre em Required variables e defina:
ibrav: Free lattice
celldm(1): 1.889725989
nat: 3
ntyp: 1
ecutwfc: 20
ecutrho: 160
2.2.3) Na aba System entre em Optional Variables e defina:
occupation = smearing
degauss = 0.03
smearing = gaussian
2.2.4) Na aba Lattice & Atomic data:
Enter Lattice Basis Vectors
Parâmetro de rede experimental do alumínio: 4.05 Angstrom
2.025 2.025 0.00
2.025 -2.025 0.00
0.00 0.00 12.30
Enter Atomic Types
Defina a massa do alumínio como 27.0
Use o botão "Pseudopotential ..." para escolher o arquivo com o pseudopotencial Al.pw91-n-van.UPF
Enter Atomic Coordinate Unit
Selecionar angstroms
Enter Atomic Coordinate
Colocar cada átomo de alumínio e suas respectivas coordenadas X, Y e Z, sem otimização. As posições são:
Al 0.000000000 0.000000000 0.000000000
Al 2.025000000 0.000000000 2.025000000
Al 0.000000000 0.000000000 4.050000000
2.2.5) Na aba K-point data:
Selecionar "automatic"
Selecionar: nk1= 8 nk2= 8 nk3= 1
sk1= 1 sk2= 1 sk3= 1
2.3) Grave esse arquivo com o nome Al001-scf.pw.inp e abra-o no programa XCrysDen para visualizar se esta tudo correto.
2.4) Para executar a simulação:
prompt> pw.x <Al001-scf.pw.inp> Al001-scf.pw.out &
Verifique a convergência e o valor da energia total. Multiplique por três a energia total obtida para o Al na prática Primeiros Passos em Metais e compare com a energia total do slab que você acabou de calcular.
prompt> vim Al001-scf.pw.out
Uma forma de fazer o teste da camada de vácuo seria aumentar de 1 em 1 angstrom o valor referente ao parâmetro de rede c, em CELL PARAMETERS, já que o teste de camada de vácuo é para 0 0 1.
Ex: O primeiro scf tem os seguinte parâmetro de rede:
2.025 2.025 0.000
2.025 -2.025 0.000
0.000 0.000 12.300
O segundo teria:
2.025 2.025 0.000
2.025 -2.025 0.000
0.000 0.000 13.300
O terceiro teria:
2.025 2.025 0.000
2.025 -2.025 0.000
0.000 0.000 14.300
Isto seria feito até 19.30, em que seriam criados diversos arquivos e calcularia todos eles. Ao final, construiria um gráfico de energia total x vácuo e analisaria a convergência.
4) Cálculo de relaxamento da primeira camada superficial.
4.1) Copie e edite o arquivo Al001-scf.pw.inp:
prompt> cp Al001-scf.pw.inp Al001-relax.pw.inp
prompt> vim Al001-relax.pw.inp
OBS: Mudar title e prefix em &CONTROL para Al001-relax.pw.
4.2) Alterar as seguintes partes do arquivo:
4.2.1) trocar "calculation = scf" por "calculation = relax"
4.2.2) adicionar "1 1 1" após as coordenadas do primeiro átomo (permitirá a otimização de posição)
4.2.3) adicionar "0 0 0" após as coordenadas do segundo átomo (não permitirá a otimização de posição)
4.2.4) adicionar "0 0 0" após as coordenadas do terceiro átomo (não permitirá a otimização de posição)
OBS: Adicione a aba &IONS depois da aba &ELECTRONS e salve o arquivo. Caso opte por fazer as edições acima pelo PWgui, a aba &IONS é incluída automaticamente ao salvar o arquivo.
4.4) Para executar a simulação:
prompt> pw.x <Al001-relax.pw.inp> Al001-relax.pw.out &
4.5) Verifique o resultado. Particularmente, observar os valores das posições atômicas e da energia total. Compare com a energia total do slab não relaxado.
prompt> vim Al001-relax.pw.out
10. Estados de transição (voltar ao índice)
Diretório original: /praticas-gfqsi/neb
1) Baixe o arquivo neb (Se o download falhar, copie e cole o link em uma nova aba do navegador e dê enter) e descompacte-o dentro do seu diretório de práticas.
2) Crie o diretório H2+H dentro do seu diretório neb. Copie o arquivo H.pw91-van_ak.UPF do diretório original para o diretório H2+H que você criou.
3) Com o comando vim, crie o arquivo H2+H.neb.inp dentro do diretório neb/H2+H
BEGIN
BEGIN_PATH_INPUT
&PATH
string_method = 'neb' ,
nstep_path = 1000 ,
ds = 2.D0 ,
opt_scheme = "broyden" ,
num_of_images = 7 ,
k_max = 0.3D0 ,
k_min = 0.2D0 ,
path_thr = 0.1D0 ,
/
END_PATH_INPUT
BEGIN_ENGINE_INPUT
&CONTROL
prefix = "H2+H.neb" ,
outdir = "/home/usuario/praticas/neb/H2+H" ,
pseudo_dir = "/home/usuario/praticas/neb/H2+H" ,
/
&SYSTEM
ibrav = 0 ,
celldm(1) = 1.D0 ,
nat = 3 ,
ntyp = 1 ,
ecutwfc = 20.0D0 ,
ecutrho = 100.0D0 ,
nspin = 2 ,
starting_magnetization = 0.5D0 ,
occupations = "smearing" ,
degauss = 0.03D0 ,
/
&ELECTRONS
conv_thr = 1.D-8 ,
mixing_beta = 0.3D0 ,
electron_maxstep = 1000 ,
/
&IONS
ds = 2.D0 ,
opt_scheme = 'broyden' ,
num_of_images = 7 ,
k_max = 0.3D0 ,
k_min = 0.2D0 ,
CI_scheme = 'no-CI' ,
pot_extrapolation = 'second_order' ,
wfc_extrapolation = 'second_order' ,
path_thr = 0.1D0 ,
/
CELL_PARAMETERS cubic
12.0 0.0 0.0
0.0 5.0 0.0
0.0 0.0 5.0
ATOMIC_SPECIES
H 1.00794 H.pw91-van_ak.UPF
BEGIN_POSITIONS
FIRST_IMAGE
ATOMIC_POSITIONS { bohr }
H -4.566700090 0.0 0.0 1 0 0
H 0.0 0.0 0.0 0 0 0
H 1.557766760 0.0 0.0 1 0 0
LAST_IMAGE
ATOMIC_POSITIONS { bohr }
H -1.557766760 0.0 0.0
H 0.0 0.0 0.0
H 4.566700090 0.0 0.0
END_POSITIONS
END_ENGINE_INPUT
END
OBS: Não se esqueça de trocar a palavra usuario pelo seu nome de usuario em outdir e pseudo_dir.
4) Para rodar o cálculo de neb, deve-se usar o seguinte comando:
prompt> neb.x -inp H2+H.neb.inp > H2+H.neb.out &
Ao terminar o cálculo visualize o arquivo gerado H2+H.axsf com o XCrysDen.
Dicas importantes para o estudo do estado de transição.
O caminho de transição é constituído por um conjunto de imagens, cuja quantidade é indicada no arquivo de entrada através da
variável “num_of_images”. Antes de qualquer cálculo é possível verificar todas as imagens com o auxílio do xcrysden. Por exemplo:
prompt> xcrysden &
abrir o arquivo de entrada (“Open PWscf” – “Open Pwscf Input file” – “seu arquivo de entrada”), deste modo você poderá ver as imagens propostas. Este procedimento é importante para verificar possíveis erros ou inconsistências. Por exemplo:
1) Copie o arquivo “agua_MgO-err.neb.inp” do diretório original.
2) Verifique as imagens no xcrysden.
Observe que as imagens propostas entre o reagente e produto não são adequadas.
3) Agora leia o arquivo.
prompt> vim agua_MgO-err.neb.inp
Note que a ordem dos atómos listados na aba “ATOMIC_POSITIONS” do reagente não é a mesma que a do produto, e é justamente esta discondância a responsável pela desordem observada nas imagens propostas. Reordenar os átomos resolve a maior parte ou totalmente o problema, como no exemplo mostrado a seguir:
1) Copie o arquivo “agua_MgO-ok.neb.inp”.
2) Verifique as imagens no xcrysden.
Observe que as imagens propostas neste caso são muito mais coerentes que a anterior. Logo, este cuidado deve ser tomado sempre.
Dissociação de H2O na superfície da brucita
1) Baixe o arquivo H2O/brucita (Se o download falhar, copie e cole o link em uma nova aba do navegador e dê enter) e descompacte-o dentro do seu diretório de práticas.
2) Abra o arquivo bruH2Odisneb.inp no Xcrysden;
3) Visualize o caminho de reação através da janela do Animation Control Center
4) Abra o arquivo bruH2Odisneb.inp no terminal e verifique como é um arquivo neb e as variáveis que são necessárias para rodar o neb;
5) Faça as devidas modificações em outdir e pseudo_dir;
Repare que foram incluídas algumas imagens intermediárias, geralmente só a primeira e a última imagens são necessárias. Estas imagens foram adicionadas para que o cálculo rode rápido. Além disso, as coordenadas deste arquivo já se encontram otimizadas e o caminho também, assim o cálculo será bem rápido. Entretanto, um cálculo neb pode demorar muitos dias, semanas e até meses, isto vai depender da complexidade do sistema.
Rode o cálculo com o comando:
prompt> neb.x -inp bru-H2Odis-neb.inp > bru-H2Odis.out &
Verifique os arquivos gerados:
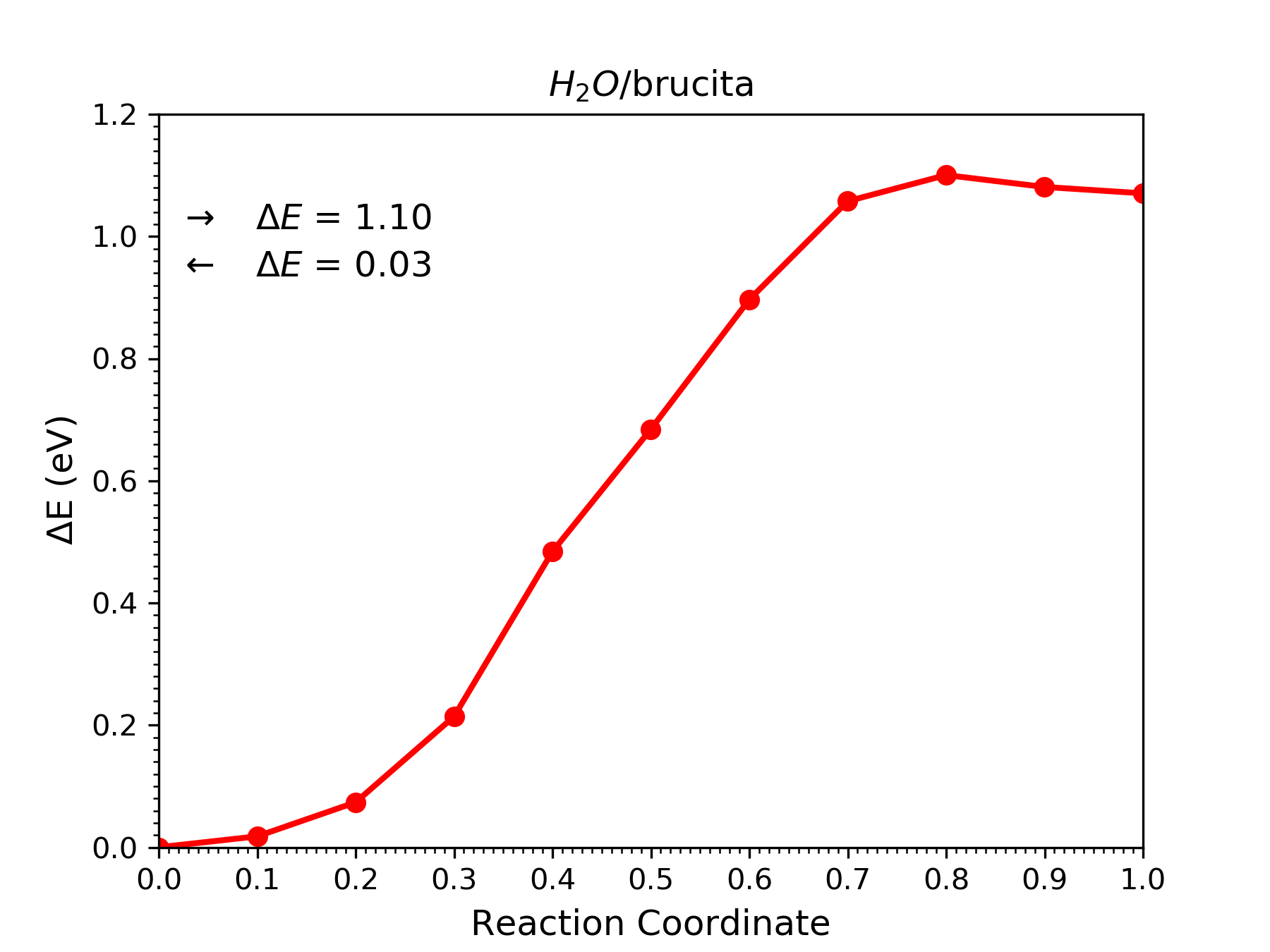
– O arquivo bru-H2Odis-neb.out contém as etapas de otimização do caminho, as energias de cada imagem, o erro com relação a convergência da força do caminho, as energias de ativação da reação direta e inversa e indica qual imagem é o estado de transição;
– Os arquivos pw_1.in, pw_2.in, … , são arquivos iguais os utilizados para rodar um cálculo relax;
– O arquivo bru-H2Odis-neb.xyz possui as coordenadas atuais (etapa na qual o cálculo se encontra) de todos os átomos de todas as imagens;
– O arquivo bru-H2Odis-neb.dat possui coordenada de reação, a energia relativa de cada imagem com relação a primeira e o erro (da etapa atual). Pode ser usado para fazer a figura com os pontos do caminho de reação;
– O arquivo bru-H2Odis-neb.axsf permite visualizar o caminho de reação atual no Xcrysden;
– O arquivo bru-H2Odis-neb.path contém informações para restartar o cálculo como as coordenadas dos átomos de cada imagem, as energias e os gradientes;
Verifique o diretório outdir:
– Cada diretório bru-H2Odis-neb_1, … possui todos os arquivos gerados num cálculo relax inclusive o arquivo bru-H2Odis-neb.out;
– Cada arquivo bru-H2Odis-neb.path possui as informações para restartar o cálculo;
Construa o gráfico com o caminho de reação.
No terminal abra o xmgrace, na aba Data clique em Import e depois em ASCII, encontre o arquivo bru-H2Odis-neb.dat e abra-o. O gráfico obtido deve ter um aspecto semelhante ao observado nesta figura.
{kind=link}
11. Pseudopotenciais (voltar ao índice)
Construindo um PP
1) O 1º passo para a geração de um pseudopotencial é fazer um cálculo com todos os elétrons. O silício será usado como exemplo.
&INPUT
iswitch = 1 , #cálculo com todos os elétrons
atom = 'Si' ,
config = '[Ne] 3s2.0 3p2.0' ,
rel = 1 , #cálculo Relativístico Escalar (o comum)
dft = 'PBE' , #funcional de troca-correlação
title = 'silicio' , #opcional
prefix = 'Si.pbe' , #também opcional
/
2) Retire os comentários, salve o arquivo como si_ae.inp e o execute com o ld1.x:
prompt> ld1.x < si_ae.inp > si_ae.out
Será gerado um arquivo Si.pbe.wfc que pode ser visualizado com o programa xmgrace.
3) Deve-se modificar o arquivo Si.pbe.wfc, deletando a primeira linha e renomeando para Si_editado_PBE.dat. Logo após, digite no terminal o comando xmgrace, importe o arquivo dat como load as NXY.
4) Visualize o gráfico e observe o comportamento radial dos orbitais.
5) O 2º passo é calcular as derivadas logarítmicas. Estas serão particularmente importantes para testar o pseudopotencial gerado à procura de estados “fantasmas”. Crie o arquivo si_ae_ld.inp como abaixo:
&INPUT
iswitch = 1 , #cálculo com todos os elétrons
atom = 'Si' ,
config = '[Ne] 3s2.0 3p2.0' ,
rel = 1 , #cálculo Relativístico Escalar (o comum)
dft = 'PBE' , #funcional de troca-correlação
title = 'silicio' , #opcional
prefix = 'Si.pbe' , #também opcional
nld = 3 , #derivadas logarítmicas s,p,d
rlderiv = 2.1 , #raio em que as derivadas logarítmicas serão calculadas
eminld = -11.0 , #energia mínima das derivadas logarítmicas
emaxld = 2.0 , #energia máxima das derivadas logarítmicas
deld = 0.01 , #intervalo de energia em que as derivadas são calculadas (em Ry)
/
6) Retire os comentários, salve o arquivo e o execute com o ld1.x. Será criado o arquivo Si.pbe.dlog. Seu formato é: energias na primeira coluna, derivadas logarítmicas s, p, d… nas seguintes.
7) Faça o mesmo procedimento no programa xmgrace.
8) O 3º passo é, enfim, gerar o pseudopotencial. Crie o arquivo si_ps.inp como segue abaixo:
&INPUT
iswitch = 3 , #geração de pseudopotencial
atom = 'Si' ,
config = '[Ne] 3s2.0 3p2.0 3d-1' , #repare a inclusão do 3d como canal local
rel = 1 ,
dft = 'PBE' ,
title = 'silicio' ,
prefix = 'Si.pbe' ,
nld = 3 ,
rlderiv = 2.1 ,
eminld = -11.0 ,
emaxld = 2.0 ,
deld = 0.01 ,
/
&InputP
pseudotype = 3 , #pseudopotencial ultrasoft
file_pseudopw = 'Si.pbe.UPF' , #nome do arquivo que conterá o pseudopotencial gerado
author = 'seu nome' #opcional
lloc = 2 , #momentum angular do canal local (no caso, d)
zval = 4.0 , #número de elétrons da valência
rho0 = 0.01 , #carga na origem
/
5 #número de estados a serem pseudizados
3S 1 0 2.000 0.000 2.00 2.50 #"orbital" "N do orbital dentro do pseudo" "L do orbital" "Ocupação" "Energia do estado" "R_cut" "R_us"
3S 1 0 0.000 -0.300 2.00 2.50
3P 2 1 2.000 0.000 2.10 2.50
3P 2 1 0.000 -0.100 2.10 2.50
3D 3 2 0.000 -0.100 2.50 2.50
OBS: Antes de rodar o cálculo, algumas observações precisam ser feitas:
(1) Pseudopotenciais ultrasoft (Vanderbilt) requerem mais de um canal por momento angular para atender a uma boa condição de transferabilidade. Daí a inclusão de canais 3s e 3p vazios e pseudizados em energias negativas: -0,3 Ry e -0,1 Ry, respectivamente. Os valores negativos na energia destes canais significa que eles não são ligantes no estado fundamental. Tais energias são escolhidas de forma mais ou menos arbitrária no intervalo de energia típico para elétrons de valência. Se ambas as energias fossem -0,1 Ry, isso estaria muito longe do estado ligante e o algoritmo não conseguiria produzir uma solução sem nós (a pseudofunção de onda não pode conter nós). Se ambas fossem -0,3 Ry e a carga na origem fosse zero (rho0 = 0), também não iria funcionar. Com um valor não nulo de carga na origem, força-se uma forma diferente na pseudofunção e aí funciona. Enfim, é caso de fazer tentativas…
(2) A inclusão do estado 3d (que também não é ligante no estado fundamental e é pseudizado em uma energia negativa) é devida a essa condição de transferabilidade dos pseudopotenciais ultrasoft. Como o canal local precisa ser único e é necessário ter mais de um canal para os estados ligantes, o primeiro estado não-ligante (neste caso, o 3d) foi incluso para ser o canal local.
(3) O valor “0,00” como energia significa que aqueles estados serão pseudizados na energia de Kohn-Sham do estado ligante (que é conhecida através do cálculo all-electron).
(4) Assume-se a técnica de Rabe-Rappe-Kaxiras-Joannopoulos (RRKJ) para a “pseudização”.
(5) É necessário definir os raios de corte ultrasoft (R_US) maiores que os raios de corte para o caso de norma conservada.
(6) O canal local tem que ser o último da lista.
9) Agora remova os comentários e rode o cálculo com o ld1.x.
10) Apague a primeira linha do arquivo Si.pbeps.wfce salve com outro nome no formato .dat.
11) Abra-o no programa xmgrace como load_as NXY. Repare que as curvas relativas ao estados 3s (linhas pretas) e 3p (linhas vermelhas) da função de onda real e da pseudofunção de onda coincidem a partir do raio de corte. Note também que as pseudofunções de onda não têm nós antes do raio de corte.
12) Repita o procedimento para as derivadas logarítmicas, abrindo o arquivo Si.pbeps.dlog no programa xmgrace. O teste das derivadas logarítmicas é importante para avaliar se o pseudopotencial gerado não possui estados “fantasmas”, isto é, nós abaixo da energia de referência. As derivadas logarítmicas oscilam bruscamente nos nós. A função de onda real destes estados possui nós. Estes não aparecem aqui porque o intervalo de energia escolhido (eixo x) é relevante para a valência, onde estes estados não tem nós. A pseudofunção de onda não pode ter nós em lugar algum.
13) Embora o pseudopotencial já tenha sido gerado, ele ainda precisa ser testado. Para tanto, crie o seguinte arquivo:
&INPUT
iswitch = 2 ,
atom = 'Si' ,
config = '[Ne] 3s2.0 3p2.0 3d-1' ,
rel = 1 ,
dft = 'PBE' ,
title = 'silicio' ,
prefix = 'Si.pbe' ,
/
&test
file_pseudo="Si.pbe.UPF",
nconf=5,
configts(1)="3s2 3p2",
configts(2)="3s2 3p1.5 3d0",
configts(3)="3s1 3p3",
configts(4)="3s1 3p2",
configts(5)="3s2 3p1 3d0",
/
14) Salve-o como si_test.inp e execute-o com o ld1.x. Um resumo dos resultados sairá no arquivo Si.pbe.test. Repare que as configurações com ocupações fracionárias são aceitas! Fisicamente, pode-se pensar que um átomo com essa configuração está parcialmente ionizado. Repare ainda que as pseudo-energias não têm significado físico, pois dependem do pseudopotencial. Apenas diferenças de energia são relevantes.
15) Verifique as diferenças de cada configuração, abrindo os arquivos Si.pbe1ps.wfc, Si.pbe2ps.wfc, Si.pbe3ps.wfc, Si.pbe4ps.wfc e Si.pbe5ps.wfc e apagando a primeira linha. Abra-os no programa xmgrace.
Correção não-linear de caroço (NLCC)
1) Para gerar um pseudopotencial com Correção Não-Linear de Caroço, basta incluir a variável nlcc na aba &InputP do arquivo de geração do PP (3º passo), como descrito abaixo:
&INPUT
iswitch = 3 ,
atom = 'Si' ,
config = '[Ne] 3s2.0 3p2.0 3d-1' ,
rel = 1 ,
dft = 'PBE' ,
title = 'silicio' ,
prefix = 'Si.pbe_nlcc' ,
nld = 3 ,
rlderiv = 2.1 ,
eminld = -11.0 ,
emaxld = 2.0 ,
deld = 0.01 ,
/
&InputP
pseudotype = 3 ,
file_pseudopw = 'Si.pbe_nlcc.UPF' ,
author = 'seu nome'
lloc = 2 ,
zval = 4.0 ,
rho0 = 0.01 ,
nlcc = .true. ,
/
5
3S 1 0 2.000 0.000 2.00 2.50
3S 1 0 0.000 -0.300 2.00 2.50
3P 2 1 2.000 0.000 2.10 2.50
3P 2 1 0.000 -0.100 2.10 2.50
3D 3 2 0.000 -0.100 2.50 2.50
O restante do procedimento continua o mesmo.
PWA
1) Para construir um [PAW], basta incluir a variável lpaw na aba &InputP do arquivo de geração de PP (3º passo), como descrito abaixo:
&INPUT
iswitch = 3 ,
atom = 'Si' ,
config = '[Ne] 3s2.0 3p2.0 3d-1' ,
rel = 1 ,
dft = 'PBE' ,
title = 'silicio' ,
prefix = 'Si.pbe' ,
nld = 3 ,
rlderiv = 2.1 ,
eminld = -11.0 ,
emaxld = 2.0 ,
deld = 0.01 ,
/
&InputP
pseudotype = 3 ,
file_pseudopw = 'Si.PAW.UPF' ,
author = 'seu nome'
lloc = 2 ,
zval = 4.0 ,
rho0 = 0.01 ,
lpaw = .true. ,
/
5
3S 1 0 2.000 0.000 2.00 2.50
3S 1 0 0.000 -0.300 2.00 2.50
3P 2 1 2.000 0.000 2.10 2.50
3P 2 1 0.000 -0.100 2.10 2.50
3D 3 2 0.000 -0.100 2.50 2.50
O restante do procedimento continua o mesmo.